Powered by Techstars
Powered by Techstars
Post-Market Workflows
Automated for MedTech
Post-Market Workflows
Automated
for MedTech
AI-Enabled Quality and Regulatory Workflows.
FDA 21 CFR Part 11 Compliant
Supports MDR & IVDR workflows
End-to-end audit trails
FDA 21 CFR Part 11 Compliant
Supports MDR & IVDR workflows
End-to-end audit trails

Built for Medtech. Keep post-market work under control
Manage complaints, investigations, and vigilance activities in one place
Apply consistent severity, risk, and reportability criteria
Track trends across products, time, and regions to surface emerging signals
Turn ongoing work into audit-ready reports
Connect PMS data, supporting evidence, and decisions as they evolve
Capture literature review and performance evidence preserved citations and rationale
Support PSUR, PMPF, CER and other MDR/IVDR post-market reporting workflows
Stay current as regulatory requirements evolve, including MDR/IVDR
Monitor updates to regulations, guidance, and standards across regions
See which products, documents, and reports are impacted
Maintain a traceable record of what changed, what was reviewed, and what was updated
Your regulatory workflows, connected in one system.
What the Cytodyme Platform Supports:
Built for Medtech. Keep post-market work under control
Manage complaints, investigations, and vigilance activities in one place
Apply consistent severity, risk, and reportability criteria
Track trends across products, time, and regions to surface emerging signals


Turn ongoing work into audit-ready reports
Connect PMS data, supporting evidence, and decisions as they evolve
Capture literature review and performance evidence preserved citations and rationale
Support PSUR, PMPF, CER and other MDR/IVDR post-market reporting workflows
Stay current as regulatory requirements evolve, including MDR/IVDR
Monitor updates to regulations, guidance, and standards across regions
See which products, documents, and reports are impacted
Maintain a traceable record of what changed, what was reviewed, and what was updated

Built for the way regulatory and quality teams work
Replace repetitive reporting and disconnected files with AI-powered automation built for quality, regulatory, and post-market teams.
Post-Market Reporting
Post-Market Reporting
Automatically generate post-market and submission-ready reports from connected, traceable source data, including PSUR, PMPF, CER, MDR reports, and other required post-market outputs.

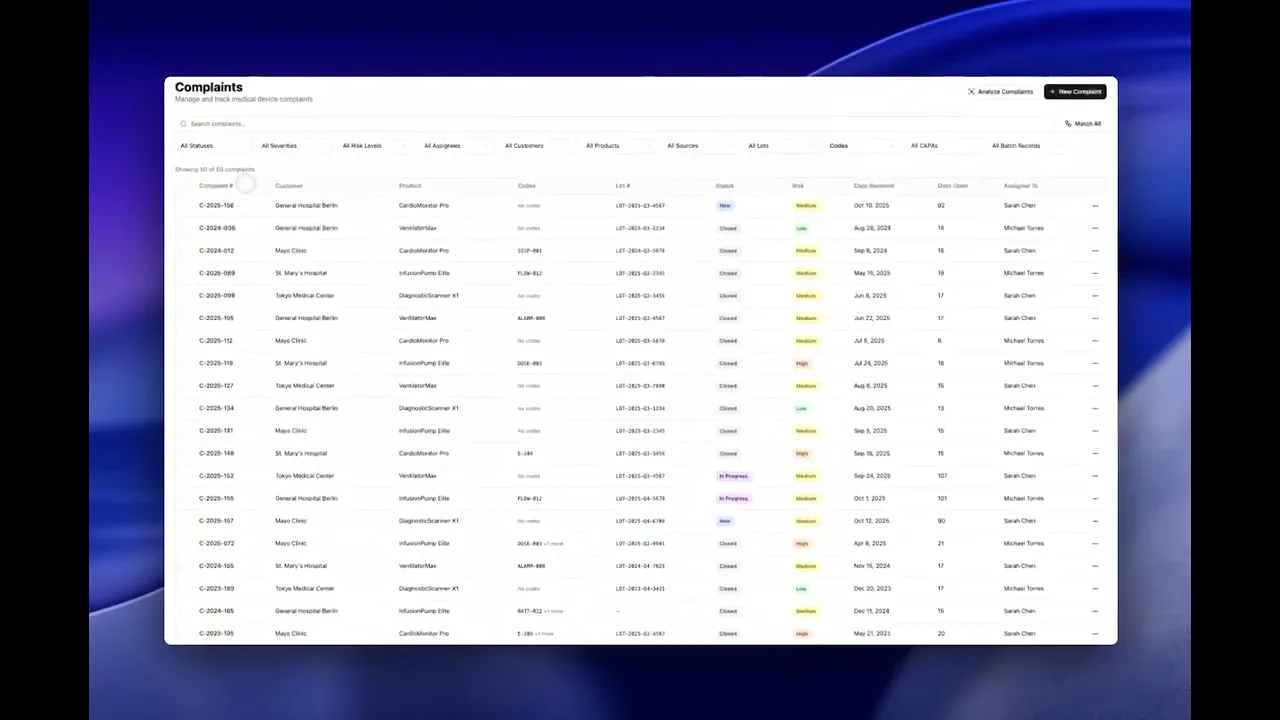
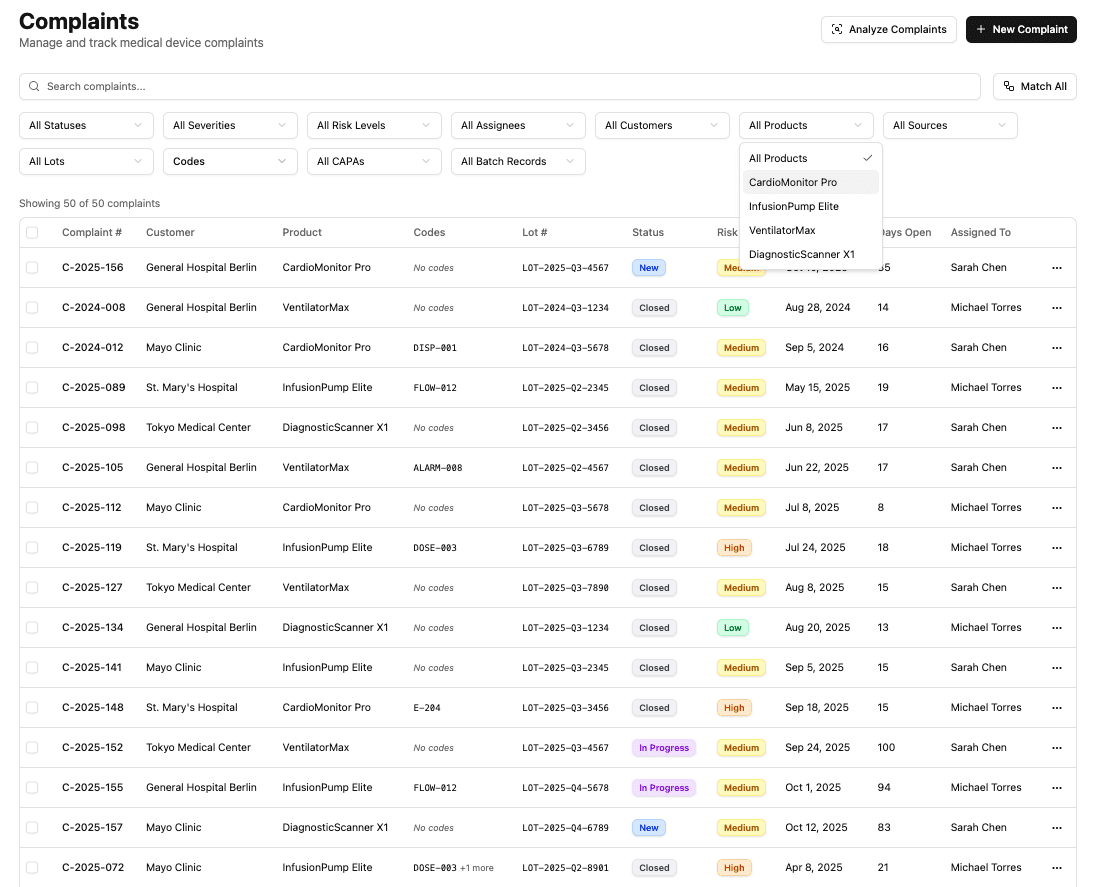
Complaint Management
Complaint Management
Complaint Management
Capture, assess, and act on product issues before they escalate. Centralize complaints from every intake channel, assess patient risk and reportability, and manage investigations in a single system.
Literature Surveillance [PMS]
Literature Surveillance [PMS]
Literature Surveillance [PMS]
Evidence monitoring at the scale regulators expect. Run validated literature searches, screen evidence at scale, and produce audit-ready outputs for clinical evaluation and post-market obligations.

Lean Quality Management System [Q]
Lean Quality Management System [Q]
Quality Management System [Q]
A modern approach to a QMS, designed to evolve alongside product changes, regulatory updates, and operational workflows.

Pain-free complaint intake
Automatically group similar issues, even when written differently.
Pain-free complaint intake
Automatically group similar issues, even when written differently.
Risk that updates as you learn
Severity and patient impact adjust as new facts appear
Risk that updates as you learn
Severity and patient impact adjust as new facts appear
Surface siloed data
Track UDI, lot, serial, and product history together
Surface siloed data
Track UDI, lot, serial, and product history together
Clear reportability decisions
Document why an event is reportable or not
Clear reportability decisions
Document why an event is reportable or not
Faster regulatory reporting
Draft MDR, IVDR, and FDA reports from live data
Faster regulatory reporting
Draft MDR, IVDR, and FDA reports from live data
Continuous literature review
Literature screening that runs continuously in the background
Continuous literature review
Literature screening that runs continuously in the background
Audit trails without extra work
Every change, decision, and approval automatically recorded
Audit trails without extra work
Every change, decision, and approval automatically recorded
Validated for compliance
Designed to support EMA/FDA 21 CFR Part 11 requirements
Validated for compliance
Designed to support EMA/FDA 21 CFR Part 11 requirements
Pain-free complaint intake
Automatically group similar issues, even when written differently.
Risk that updates as you learn
Severity and patient impact adjust as new facts appear
Surface siloed data
Track UDI, lot, serial, and product history together
Clear reportability decisions
Document why an event is reportable or not
Faster regulatory reporting
Draft MDR, IVDR, and FDA reports from live data
Continuous literature review
Literature screening that runs continuously in the background
Audit trails without extra work
Every change, decision, and approval automatically recorded
Validated for compliance
Designed to support EMA/FDA 21 CFR Part 11 requirements
Real Feedback. Real Teams.
What Quality and Regulatory Leaders are Saying: After seeing a better way
See what Industry Insiders are saying about Cytodyme
Vice President of Quality
Medical Device Company
Post-market requirements are growing faster than our systems can support. This meaningfully reduces the documentation burden.
QA/RA Director
Early-Stage MedTech
Documentation used to take up almost all of my time. This changes how a small team operates.
Senior Director of Quality
Medical Device Company
Ten to fifteen hours a week go toward documentation alone. Reducing that workload makes a real difference.
Principal Quality Engineer
Post-Market Risk, Enterprise MedTech
Teams are still relying on Word and Excel to manage enormous complaint volumes. That’s not sustainable.
Director of QARA Program Strategy
Surgical Devices Company
This supports how commercial quality and post-market surveillance actually work today.
Post-Market Surveillance Specialist
Orthopedic Device Manufacturer
I’d rather spend my time analyzing conclusions than pulling information together. This shifts the focus to insight.
Director of Regulatory Affairs
Diagnostic Medical Device Company
One report can take days or even a week. Faster generation changes how we plan and prioritize work.
Quality & Regulatory Leader
Medical Device Company
The hardest part isn’t knowing what to do. It’s managing the volume, traceability, and review.
Director of Quality
Medical Device Company
Traceability across complaints, reports, and risk files is one of the hardest parts of the job. Having that connected in one place changes everything.
Real Feedback. Real Teams.
What Quality and Regulatory Leaders are Saying: After seeing a better way
See what Industry Insiders are saying about Cytodyme
Post-Market Surveillance Specialist
Orthopedic Device Manufacturer
I’d rather spend my time analyzing conclusions than pulling information together. This shifts the focus to insight.
Director of Regulatory Affairs
Diagnostic Medical Device Company
One report can take days or even a week. Faster generation changes how we plan and prioritize work.
Quality & Regulatory Leader
Medical Device Company
The hardest part isn’t knowing what to do. It’s managing the volume, traceability, and review.
Director of Quality
Medical Device Company
Traceability across complaints, reports, and risk files is one of the hardest parts of the job. Having that connected in one place changes everything.
Vice President of Quality
Medical Device Company
Post-market requirements are growing faster than our systems can support. This meaningfully reduces the documentation burden.
QA/RA Director
Early-Stage MedTech
Documentation used to take up almost all of my time. This changes how a small team operates.
Senior Director of Quality
Medical Device Company
Ten to fifteen hours a week go toward documentation alone. Reducing that workload makes a real difference.
Principal Quality Engineer
Post-Market Risk, Enterprise MedTech
Teams are still relying on Word and Excel to manage enormous complaint volumes. That’s not sustainable.
Director of QARA Program Strategy
Surgical Devices Company
This supports how commercial quality and post-market surveillance actually work today.
Real Feedback. Real Teams.
What Quality and Regulatory Leaders are Saying: After seeing a better way
See what Industry Insiders are saying about Cytodyme
Director of Quality
Medical Device Company
Traceability across complaints, reports, and risk files is one of the hardest parts of the job. Having that connected in one place changes everything.
Director of Regulatory Affairs
Diagnostic Medical Device Company
One report can take days or even a week. Faster generation changes how we plan and prioritize work.
Senior Director of Quality
Medical Device Company
Ten to fifteen hours a week go toward documentation alone. Reducing that workload makes a real difference.
Vice President of Quality
Medical Device Company
Post-market requirements are growing faster than our systems can support. This meaningfully reduces the documentation burden.
Post-Market Surveillance Specialist
Orthopedic Device Manufacturer
I’d rather spend my time analyzing conclusions than pulling information together. This shifts the focus to insight.
QA/RA Director
Early-Stage MedTech
Documentation used to take up almost all of my time. This changes how a small team operates.
Principal Quality Engineer
Post-Market Risk, Enterprise MedTech
Teams are still relying on Word and Excel to manage enormous complaint volumes. That’s not sustainable.
Quality & Regulatory Leader
Medical Device Company
The hardest part isn’t knowing what to do. It’s managing the volume, traceability, and review.
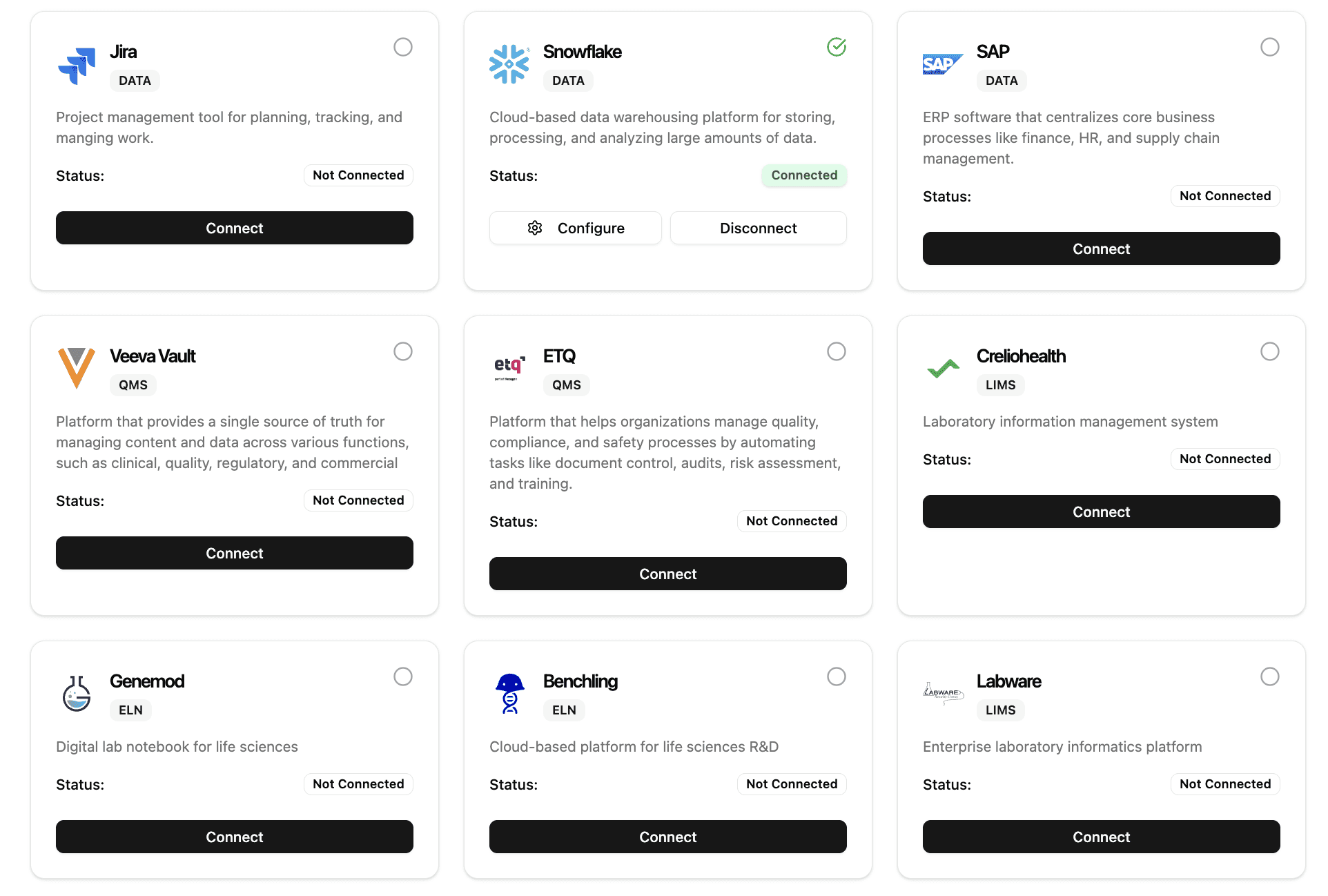
Works with your favorite tools
We play nice with others:
Connect to your tool stack in seconds.
Integrates with existing quality, safety, and data sources to maintain continuity across workflows.
Works with your favorite tools
We play nice with others:
Connect to your tool stack in seconds.
Integrates with existing quality, safety, and data sources to maintain continuity across workflows.
Works with your favorite tools
We play nice with others:
Connect to your tool stack in seconds.
Integrates with existing quality, safety, and data sources to maintain continuity across workflows.
Continuously Meet & Maintain Compliance Standards
Receive guidance, change signals, and structured outputs to help teams stay aligned with standards including:
ISO 13485
ISO 13485
ISO 14971
ISO 14971
ISO 17025
ISO 17025
ISO 27001
ISO 27001
ISO 9001
ISO 9001
ISO 15189
ISO 15189
FDA 21 CFR Part 11
FDA 21 CFR Part 11
FDA 21 CFR Part 4
FDA 21 CFR Part 4
EU/UK MDR
EU/UK MDR
EU IVDR
EU IVDR
GDPR
GDPR
CAP
CAP
ISO 13485
ISO 14971
ISO 17025
ISO 27001
ISO 9001
ISO 15189
FDA 21 CFR Part 11
FDA 21 CFR Part 4
EU/UK MDR
EU IVDR
GDPR
CAP
Maximum Security
Cytodyme meets industry expectations for security, data integrity, and system controls, with capabilities aligned to standards such as FDA 21 CFR Part 11, ISO 13485, ICH Q10, and 21 CFR Part 820
Validation-ready Infrastructure
Built to support validation activities for quality and post-market workflows, with system controls aligned to FDA 21 CFR Part 11, ISO 13485, MDR, and IVDR expectations. Structured records, traceability, version control, and good documentation practices support inspections and audits.
Auditability & Access Control
Every action is logged, time-stamped, and attributable to a user. Role-based access controls enforce least-privilege permissions, while complete version history, approvals, and audit trails support inspection readiness and internal governance.
Security by design
Encryption at rest and in transit, OAuth 2.0, and two-factor authentication. Customer data is isolated, never used beyond its intended purpose, and AI features do not train on customer content. Supports strict data ownership and separation requirements.
Questions?
Questions?
Questions?
Additional questions? We’ve got you covered.
Who is Cytodyme built for?
Cytodyme helps medtech teams manage post-market work including complaint handling, literature surveillance, regulatory report generation, and documentation in a single connected system. By keeping data structured, up to date, and aligned with current regulatory requirements, Cytodyme reduces manual work, supports compliance, and helps teams prepare accurate reports with confidence.
What post-market workflows does Cytodyme support today?
Today, Cytodyme supports complaint management and post-market surveillance workflows, including intake, triage, documentation, literature surveillance, and report generation. The platform is designed to reduce manual effort while maintaining traceability and audit readiness.
How does Cytodyme fit into existing quality systems?
Cytodyme is built to complement existing systems, not replace them. Many teams use it alongside their current eQMS or document management tools to streamline post-market workflows, improve traceability, and reduce time spent assembling reports.
How does Cytodyme ensure regulatory compliance?
Cytodyme is purpose-built for regulated post-market workflows in medical devices and diagnostics. The platform is designed to support FDA requirements, EU MDR expectations, and ISO 13485 principles through structured workflows, version control, complete audit trails, and end-to-end traceability across complaints, reports, and supporting evidence. All outputs are generated in a review-ready, explainable format that supports regulatory justification and inspection readiness, rather than opaque, black-box automation.
Is there a human-in-the-loop review process?
Yes. Cytodyme’s goal is to remove the manual, repetitive work that slows teams down, such as assembling data, structuring documentation, and generating draft reports. The platform automates these workflows and prepares review-ready outputs, but final review, approval, and sign-off always remain with the human expert. This ensures teams move faster without sacrificing regulatory judgment or accountability.
How does Cytodyme handle data security and confidentiality?
Data security is a core priority. Cytodyme uses secure infrastructure, access controls, and role-based permissions to protect sensitive information. Customer data is never used to train models, and all workflows are designed with confidentiality in mind.
How long does it take to get started?
Most teams can get started quickly. Cytodyme is designed to deploy in days, not months, with guided onboarding and support to help teams integrate the platform into their existing workflows without disruption.
How does Cytodyme scale as requirements grow?
Cytodyme is built to scale with increasing post-market demands. As reporting frequency, product lines, or regulatory obligations grow, the platform helps teams manage higher volume without adding proportional headcount.
Who is Cytodyme built for?
Cytodyme helps medtech teams manage post-market work including complaint handling, literature surveillance, regulatory report generation, and documentation in a single connected system. By keeping data structured, up to date, and aligned with current regulatory requirements, Cytodyme reduces manual work, supports compliance, and helps teams prepare accurate reports with confidence.
What post-market workflows does Cytodyme support today?
Today, Cytodyme supports complaint management and post-market surveillance workflows, including intake, triage, documentation, literature surveillance, and report generation. The platform is designed to reduce manual effort while maintaining traceability and audit readiness.
How does Cytodyme fit into existing quality systems?
Cytodyme is built to complement existing systems, not replace them. Many teams use it alongside their current eQMS or document management tools to streamline post-market workflows, improve traceability, and reduce time spent assembling reports.
How does Cytodyme ensure regulatory compliance?
Cytodyme is purpose-built for regulated post-market workflows in medical devices and diagnostics. The platform is designed to support FDA requirements, EU MDR expectations, and ISO 13485 principles through structured workflows, version control, complete audit trails, and end-to-end traceability across complaints, reports, and supporting evidence. All outputs are generated in a review-ready, explainable format that supports regulatory justification and inspection readiness, rather than opaque, black-box automation.
Is there a human-in-the-loop review process?
Yes. Cytodyme’s goal is to remove the manual, repetitive work that slows teams down, such as assembling data, structuring documentation, and generating draft reports. The platform automates these workflows and prepares review-ready outputs, but final review, approval, and sign-off always remain with the human expert. This ensures teams move faster without sacrificing regulatory judgment or accountability.
How does Cytodyme handle data security and confidentiality?
Data security is a core priority. Cytodyme uses secure infrastructure, access controls, and role-based permissions to protect sensitive information. Customer data is never used to train models, and all workflows are designed with confidentiality in mind.
How long does it take to get started?
Most teams can get started quickly. Cytodyme is designed to deploy in days, not months, with guided onboarding and support to help teams integrate the platform into their existing workflows without disruption.
How does Cytodyme scale as requirements grow?
Cytodyme is built to scale with increasing post-market demands. As reporting frequency, product lines, or regulatory obligations grow, the platform helps teams manage higher volume without adding proportional headcount.
Who is Cytodyme built for?
Cytodyme helps medtech teams manage post-market work including complaint handling, literature surveillance, regulatory report generation, and documentation in a single connected system. By keeping data structured, up to date, and aligned with current regulatory requirements, Cytodyme reduces manual work, supports compliance, and helps teams prepare accurate reports with confidence.
What post-market workflows does Cytodyme support today?
Today, Cytodyme supports complaint management and post-market surveillance workflows, including intake, triage, documentation, literature surveillance, and report generation. The platform is designed to reduce manual effort while maintaining traceability and audit readiness.
How does Cytodyme fit into existing quality systems?
Cytodyme is built to complement existing systems, not replace them. Many teams use it alongside their current eQMS or document management tools to streamline post-market workflows, improve traceability, and reduce time spent assembling reports.
How does Cytodyme ensure regulatory compliance?
Cytodyme is purpose-built for regulated post-market workflows in medical devices and diagnostics. The platform is designed to support FDA requirements, EU MDR expectations, and ISO 13485 principles through structured workflows, version control, complete audit trails, and end-to-end traceability across complaints, reports, and supporting evidence. All outputs are generated in a review-ready, explainable format that supports regulatory justification and inspection readiness, rather than opaque, black-box automation.
Is there a human-in-the-loop review process?
Yes. Cytodyme’s goal is to remove the manual, repetitive work that slows teams down, such as assembling data, structuring documentation, and generating draft reports. The platform automates these workflows and prepares review-ready outputs, but final review, approval, and sign-off always remain with the human expert. This ensures teams move faster without sacrificing regulatory judgment or accountability.
How does Cytodyme handle data security and confidentiality?
Data security is a core priority. Cytodyme uses secure infrastructure, access controls, and role-based permissions to protect sensitive information. Customer data is never used to train models, and all workflows are designed with confidentiality in mind.
How long does it take to get started?
Most teams can get started quickly. Cytodyme is designed to deploy in days, not months, with guided onboarding and support to help teams integrate the platform into their existing workflows without disruption.
How does Cytodyme scale as requirements grow?
Cytodyme is built to scale with increasing post-market demands. As reporting frequency, product lines, or regulatory obligations grow, the platform helps teams manage higher volume without adding proportional headcount.